

| << Chapter < Page | Chapter >> Page > |
Key Concepts
In many of the physical sciences, our theoretical understanding has developed alongside experimental discoveries, for example in the fields of electromagnetism, optics and semiconductor physics. Theory has provided the design principles which have then enabled engineers to maximise the potential applications of these new technologies. However, there are many instances in which simple phenomenological models cannot capture the complexity of the systems in question: notable examples are the chemistry of the atmosphere, which has implications for prediction of weather patterns and climate change, or the properties of materials at the nanoscale, such as chemically functionalised carbon nanotubes, which will become increasingly important in nanoengineering.
Arguably, the most complex materials of all are biological macromolecules; namely proteins, DNA, lipids, sugars and their interactions. Biological macromolecules routinely perform extraordinary functions such as biomolecular recognition (Figure 1a), enzyme catalysis, self-assembly (Figure 1b) and self-organisation. Moreover, there are many examples of molecular motors within the cell (Figure 1c). These are nanoscale machines capable of burning chemical energy to perform work. The theoretical challenge of understanding these systems is more than offset by the potential benefits. For example, our current understanding of molecular recognition has already enabled us to rationally design new drugs
If we had an equivalent theoretical understanding of biological systems as we have of semiconductors, then whole new regimes of bio-inspired engineering at the nanoscale would become possible. To achieve this, we need to combine our existing physical understanding of mechanics and thermodynamics with a theoretical technique that is capable of including chemical complexity. The only suitable methodology is High Performance Supercomputing (HPC).



The most successful biomolecular simulation methods to date use Newtonian mechanics in conjunction with an empirical force-field to produce a mathematical model of the interactions between every single atom in the macromolecule with chemical accuracy; the calculation results in a series of molecular conformations (or a “movie”) that illustrates the changing shape of the biomolecule due to thermal fluctuations.
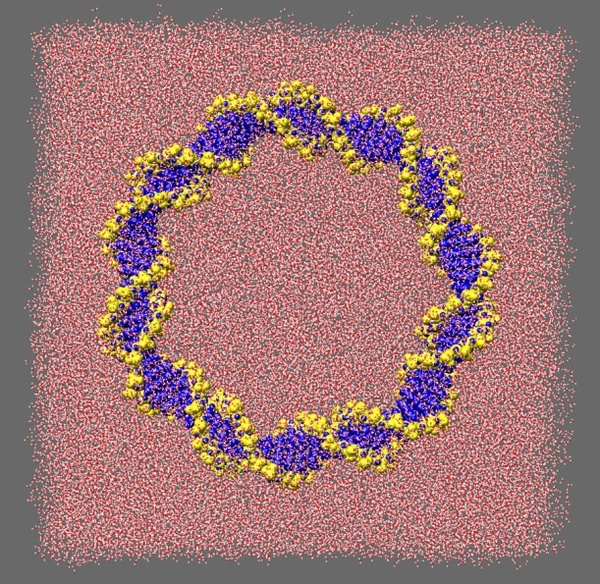
This technique is known as atomistic molecular dynamics (MD) simulation. Biomolecules are naturally highly responsive materials, as is required by their function. Consequently, the most accurate simulations of biological macromolecules must also include a description of the solvent environment (see Figure 2), which usually consists of water and counterions. Typically, such a calculation will contain ~150,000 atoms, and will require over 750 CPU hrs to obtain a 1ns MD trajectory using the AMBER suite of MD programs.
Notification Switch
Would you like to follow the 'Research in a connected world' conversation and receive update notifications?

|
|

|
|

|
|

|
|

|
|

|
|
|
|
|

|
|

|
|

|